Bond angle restraint between 3 atoms. More...
 Inheritance diagram for ObjCryst::MolBondAngle:
Inheritance diagram for ObjCryst::MolBondAngle: Collaboration diagram for ObjCryst::MolBondAngle:
Collaboration diagram for ObjCryst::MolBondAngle:
Public Member Functions | |
| MolBondAngle (MolAtom &atom1, MolAtom &atom2, MolAtom &atom3, const REAL angle, const REAL sigma, const REAL delta, Molecule &parent) | |
| Constructor. More... | |
| virtual | ~MolBondAngle () |
| Destructor. More... | |
| const Molecule & | GetMolecule () const |
| Molecule & | GetMolecule () |
| string | GetName () const |
| virtual void | XMLOutput (ostream &os, int indent=0) const |
| virtual void | XMLInput (istream &is, const XMLCrystTag &tag) |
| virtual REAL | GetLogLikelihood () const |
| Get -ln(likelihood) for this restraint. | |
| REAL | GetLogLikelihood (const bool calcDeriv, const bool recalc) const |
| REAL | GetDeriv (const std::map< const MolAtom *, XYZ > &m, const bool llk=false) const |
| Get the derivative of the angle, given the derivatives of the atom positions This requires that GetLogLikelihood(calcDeriv=true) be called first If llk=true, this will return the derivative of the llk rather than the derivative of the length or angle. | |
| void | CalcGradient (std::map< MolAtom *, XYZ > &m) const |
| Calc log(likelihood) gradient - versus all atomic coordinates. More... | |
| REAL | GetAngle () const |
| REAL & | Angle0 () |
| REAL & | AngleDelta () |
| REAL & | AngleSigma () |
| REAL | GetAngle0 () const |
| REAL | GetAngleDelta () const |
| REAL | GetAngleSigma () const |
| void | SetAngle0 (const REAL angle) |
| void | SetAngleDelta (const REAL delta) |
| void | SetAngleSigma (const REAL sigma) |
| const MolAtom & | GetAtom1 () const |
| const MolAtom & | GetAtom2 () const |
| const MolAtom & | GetAtom3 () const |
| void | SetAtom1 (MolAtom &at) |
| void | SetAtom2 (MolAtom &at) |
| void | SetAtom3 (MolAtom &at) |
| MolAtom & | GetAtom1 () |
| MolAtom & | GetAtom2 () |
| MolAtom & | GetAtom3 () |
| bool | IsFlexible () const |
| void | SetFlexible (const bool isInRing) |
| Public Member Functions inherited from ObjCryst::Restraint | |
| Restraint () | |
| Default constructor, sets RefParType to gpRefParTypeObjCryst. | |
| Restraint (const RefParType *type) | |
| constructor specifying the type | |
| virtual const RefParType * | GetType () const |
| virtual void | SetType (const RefParType *type) |
Private Attributes | |
| vector< MolAtom * > | mvpAtom |
| The vector of the 3 atoms involved in the bond angle. | |
| REAL | mAngle0 |
| REAL | mDelta |
| REAL | mSigma |
| Molecule * | mpMol |
| Parent Molecule. | |
| bool | mIsFlexible |
| When using the user-chosen flexibility model, this allows some flexibility for this bond angle, i.e. More... | |
| REAL | mLLK |
| Stored log(likelihood) | |
| XYZ | mDerivAtom1 |
| Partial derivatives of the angle with respect to the coordinates of the atoms. More... | |
| XYZ | mDerivAtom2 |
| XYZ | mDerivAtom3 |
| REAL | mDerivLLKCoeff |
| The factor used to change the derivative of the length/angle, to the derivative of the log(likelihood). More... | |
Detailed Description
Bond angle restraint between 3 atoms.
The atoms involved need not be bonded.
Definition at line 256 of file Molecule.h.
Constructor & Destructor Documentation
| ObjCryst::MolBondAngle::MolBondAngle | ( | MolAtom & | atom1, |
| MolAtom & | atom2, | ||
| MolAtom & | atom3, | ||
| const REAL | angle, | ||
| const REAL | sigma, | ||
| const REAL | delta, | ||
| Molecule & | parent | ||
| ) |
Constructor.
Definition at line 626 of file Molecule.cpp.
|
virtual |
Destructor.
Definition at line 639 of file Molecule.cpp.
Member Function Documentation
Calc log(likelihood) gradient - versus all atomic coordinates.
- Parameters
-
m this map should have been initialized by adding all possible atom pointers as keys, with all XYZ values equal to zeros. On return, the derivative of the log(likelihood) vs each atomic coordinates will added to each coordinate of the corresponding atoms.
Definition at line 863 of file Molecule.cpp.
Member Data Documentation
|
mutableprivate |
Partial derivatives of the angle with respect to the coordinates of the atoms.
The derivatives are calculated in MolBondAngle::GetLogLikelihood(true)
Definition at line 325 of file Molecule.h.
|
mutableprivate |
The factor used to change the derivative of the length/angle, to the derivative of the log(likelihood).
e.g. (for mDelta=0) 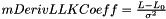
Definition at line 329 of file Molecule.h.
|
private |
When using the user-chosen flexibility model, this allows some flexibility for this bond angle, i.e.
the bond angle does not remain strictly rigid, and is still restrained.
Definition at line 318 of file Molecule.h.
The documentation for this class was generated from the following files:
